Protein function prediction using machine learning methods
We are developing machine learning-based methods to predict protein functions.
- Development of a biosynthetic gene cluster database with functional annotations
- Development of a protein functional identity prediction method
Development of a biosynthetic gene cluster database with functional annotations
The Reference Sequence Database (RefSeq) of National Center for Biotechnology Information (NCBI) registers more than 300 million protein sequences. In contrast, SwissProt, a protein function database of UniProt Knowledgebase (UniProtKB) maintained by European Bioinformatics Institute (EBI) registers only 570,000 entries of proteins with functional annotations curated by experts. In this study, we are aiming at predicting the functions of a vast number of function-unknown proteins and finding novel enzymes that catalyze useful reactions.
We have developed a database that registers the results of the function prediction and is publicly accessible via a Web server at http://sr.iu.a.u-tokyo.ac.jp/ (Fig. 1). In addition to the results of function prediction based on sequence similarity, structural models predicted by AlphaFold are available at the Web server. We will extend the database to include the function prediction results by a machine learning-based method which we are now developing.
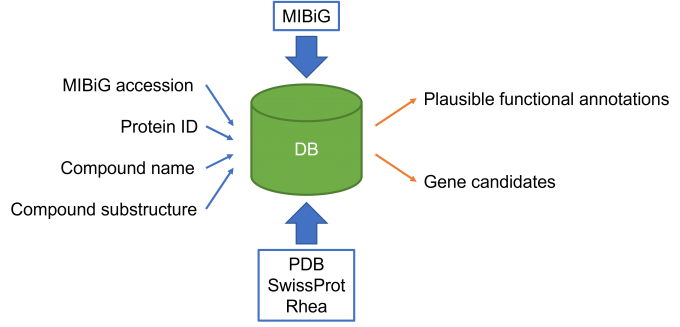 Fig. 1: Overview of the database. The database registers the results of sequence similarity search for the protein sequences in a biosynthetic gene cluster database, MIBiG, against the sequences in PDB and SwissProt. Each protein data in the database is linked to the enzymatic reaction data in Rhea if available. At the Web server, a user can search the database not only with MIBiG accession or Protein ID, but also with the name or the substructure of compound that participates in the enzymatic reaction.
Fig. 1: Overview of the database. The database registers the results of sequence similarity search for the protein sequences in a biosynthetic gene cluster database, MIBiG, against the sequences in PDB and SwissProt. Each protein data in the database is linked to the enzymatic reaction data in Rhea if available. At the Web server, a user can search the database not only with MIBiG accession or Protein ID, but also with the name or the substructure of compound that participates in the enzymatic reaction.
Development of a protein functional identity prediction method
A computational method that predicts the substrates and the products, i.e., the functions of enzymes is necessary to efficiently find novel enzymes that catalyze useful reactions from a vast number of function-unknown proteins. BLAST searches against function-known protein sequences are frequently conducted for this purpose because enzymes with similar sequences tend to have the identical function. However, their ability to detect protein sequences with the identical function is limited (see Fig. 2). We therefore developed a method to predict the functional identity of a pair of enzymes using their structural similarity in addition to their sequence similarity. Our method is termed “FUnctional identity of protein prediction by JoIning Sequence ANd structure feature (FUJISAN).” Fig. 2 shows the comparison of the prediction performance of FUJISAN with those of the BLAST E-value based method and deep-learning based methods (ESM-2 and DeepFRI). FUJISAN outperformed the other methods. The code of FUJISAN is publicly available on GitHub at https://github.com/sfujita0601/FUJISAN. We are now developing a method to comprehensively predict the functions of the biosynthetic enzymes in MIBiG using FUJISAN.
Fujita and Terada, Comput. Struct. Biotechnol. J. 23, 4124–4130 (2024).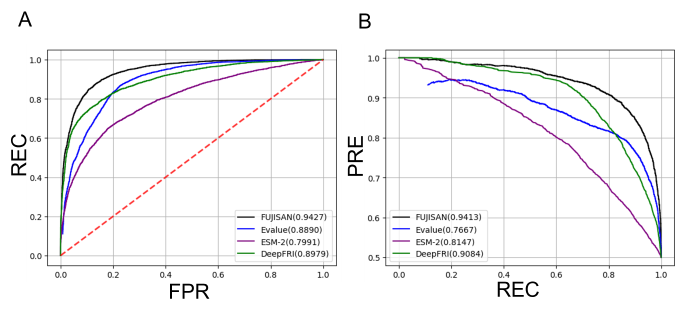 Fig. 2: Comparison of the receiver operating characteristic (ROC) curves (A) and the precision-recall (PR) curves (B) of FUJISAN, the E-value-based method, the ESM-2-based method, and the DeepFRI-based method. REC, FPR, and PRE represent recall, false positive rate, and precision, respectively. The values of the area under the ROC curves (AUROCs) and the area under the PR curves are shown in parentheses. The larger value indicates the better prediction performance.
Fig. 2: Comparison of the receiver operating characteristic (ROC) curves (A) and the precision-recall (PR) curves (B) of FUJISAN, the E-value-based method, the ESM-2-based method, and the DeepFRI-based method. REC, FPR, and PRE represent recall, false positive rate, and precision, respectively. The values of the area under the ROC curves (AUROCs) and the area under the PR curves are shown in parentheses. The larger value indicates the better prediction performance.