Molecular dynamics simulations
We conduct researches to clarify the mechanisms with which proteins exert their functions using MD simulations.
- Functional mechanism of an iron-phytosiderophore transporter
- Prediction of drug-induced blockade of cardiac ion channels
Functional mechanism of an iron-phytosiderophore transporter
An iron-phytosiderophore transporter, Yellow Stripe 1 (YS1) uptakes Fe3+ in complex with phytosiderophores (mugineic acids) and thereby plays an important role in absorbing poorly soluble iron through roots. The tertiary structures of YS1 alone and in complex with one of mugineic acids, deoxymugineic acid (DMA) (Fig. 1) binding Fe3+ (Fe(III)-DMA) were determined by cryogenic electron microscopy (cryo-EM). YS1 is a symporter that transports Fe(III)-DMA together with H+ from extracellular region to cytosol. In this study, we determined the protonation state of the cryo-EM structure and predicted the protonation sites involved in the transport. YS1 is comprised of the core domain and the scaffold domain. The transport of Fe(III)-DMA occurs via an elevator-like mechanism, in which the core domain moves with respect to the scaffold domain. We considered that some of the three acidic residues (D446, D490, and D494) located in the transmembrane regions of the core domain were involved in the transport (Fig. 2). We therefore conducted 500-ns MD simulations for YS1 with all the possible combinations of the protonation states of the three residues. Fe(III)-DMA was stably bound to the protein when only D494 was protonated, indicating that the cryo-EM structure is in this protonation state. When all the three residues were protonated, TM6 of the scaffold domain was detached from the core domain. This result suggests that the additional protonations of D446 and D490 would induce the movement of the core domain (Fig. 3). This study was conducted in collaboration with Dr. Yamagata’s group at RIKEN.
Yamagata et al., Nat. Commun. 13, 7180 (2022).
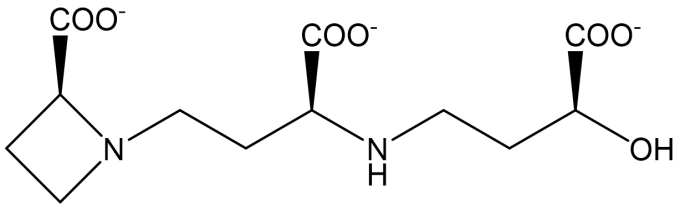
Fig. 1: The structure of deoxymugineic acid (DMA).
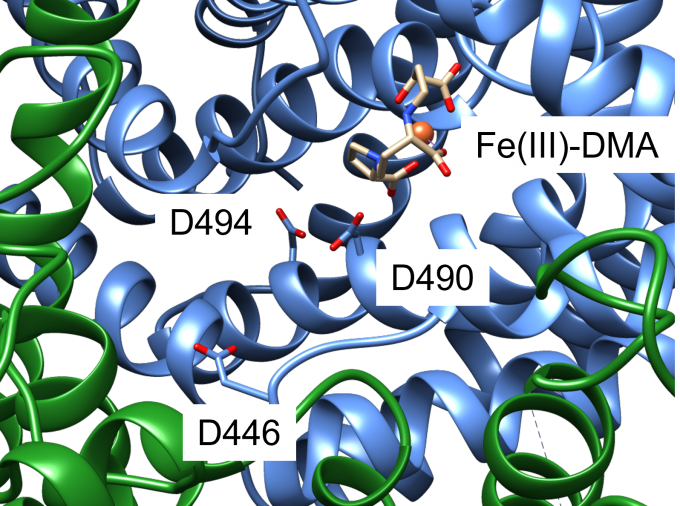
Fig. 2: A close-up view of the Fe(III)-DMA binding site of YS1. Core domain and scaffold domain are shown with blue and green ribbons, respectively. D446, D490, and D494 are shown with a stick representation. Fe3+ ion is shown with a sphere and DMA is shown with a stick representation.
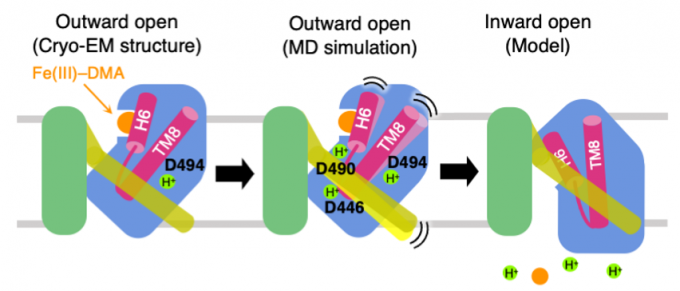
Fig. 3: Proposed mechanism of Fe(III)-DMA/H+ symport. The cryo-EM structure adopts the ourward-open conformation and stably binds Fe(III)-DMA with D494 protonated (left). When D446 and D490 are protonated in addition to D494, TM6 colored yellow is detached from the core domain colored blue (middle). Transition to the inward-open conformation occurs and Fe(III)-DMA and protons are released to the cytosol (right).
Prediction of drug-induced blockade of cardiac ion channels
A heartbeat is caused by cooperation of propagation of action potential and contraction of muscle. In the resting state, the membrane potential is polarized, i.e., the potential inside the cell is lower than outside. When the membrane potential is depolarized, Na+ channel first opens. Then, Na+ ions flow into the cell, because there are more Na+ ions outside the cell than inside. As a result, the inside potential becomes higher than the outside. This causes opening of neighboring Na+ channels and propagation of the action potential. The depolarization of the membrane potential also causes release of calcium ions from sarcoplasmic reticulum, which is a storage of calcium ions. As a result, muscle contraction occurs. On the other hand, K+ channel opens late after Na+ channel opens. Because there are more K+ ions inside the cell than outside, K+ ions flow out of the cell. This lowers the potential inside the cell and polarized membrane potential is recovered (Fig. 4). This is called repolarization. When K+ channel is blocked by a drug, the repolarization is delayed, and the QT interval of electrocardiogram is prolonged. The QT-interval prolongation may induce lethal arrhythmia. This is called cardiotoxicity, and all drug candidates are tested for cardiotoxicity in clinical trials. In the clinical trials, healthy volunteers take the drug. If the drug causes arrhythmia, development of the drug is stopped. This poses financial burden on pharmaceutical companies and raises ethical issues as well.
To alleviate these problems, computational predictions of cardiotoxicity of drugs have been attempted. In this study, we developed a method to calculate the affinities of drugs to K+ channel (hERG) and Na+ channel (NaV1.5), using their tertiary structures determined by cryogenic electron microscopy (cryo-EM). The drugs were docked into the cavity on the intracellular side of the pore in each channel structure. After refinement of the docking structures with MD simulations, the binding free energies, ∆Gbind, of the drugs to the channel were calculated with the MP-CAFEE method. The calculated ∆Gbind values of the drugs well correlated with the negative common logarithms of the experimental values of the half inhibition concentrations (pIC50) (Fig. 5). We can now predict the inhibition activity of a drug candidate against the cardiac channels by docking the drug into the channels, calculating the ∆Gbind values, and applying the calculated ∆Gbind values to the regression equations obtained here.
Negami and Terada, BPPB, 20, e200016 (2023).
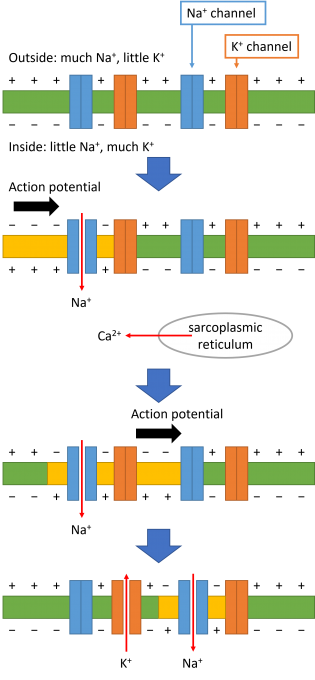
Fig. 4: Mechanism of action potential propagation.

Fig. 5: Regressions between ∆Gbind and pIC50 for Comprehensive in vitro Proarrhythmia Assay (CiPA) reference drugs. Plots for K+ channel (hERG) (left) and for Na+ channel (NaV1.5) (right).