Development and application of coarse-grained MD methods
Aiming at cell-scale simulations, we are developing coarse-grained MD simulation methods and are applying them to biological phenomena.
Coarse-grained MD simulations of protein-ligand binding processes
Because protein-ligand binding (Fig. 1) is a slow process on the time scale of milliseconds, it is not practical to reproduce this process in an all-atom MD simulation. Moreover, because protein-ligand binding is a stochastic process, the simulation must be repeated many times. In this study, we developed a method to reproduce protein-ligand binding processes using coarse-grained (CG) MD simulations.
We adopted the MARTINI model to coarse grain the protein and the ligand models. This model maps four non-hydrogen atoms to one CG bead on average. There are 18 particle types for CG beads with different interaction parameters. The particle types of an amino acid are selected so that its CG model reproduces the experimental water-oil partition coefficient of the amino acid. We choose levansucrase and sucrose for the protein and the ligand, respectively for the CGMD simulations. The ligand molecules were randomly placed around the protein. 4-μs CGMD simulations were performed 50 times with different initial ligand placement and different initial velocities. Fig. 2 shows the process of the ligand entering the substrate-binding pocket of the protein. During the CGMD simulations, 49 binding events and 18 unbinding events were observed. We calculated the binding and unbinding rate constants and the dissociation constant from these results. We found that the dissociation constant agreed well with the experimental value. The binding and unbinding rate constants were about ten timers larger than their experimental values. Considering the fact that molecules diffuse faster in a CGMD simulation due to smaller friction between them, we concluded that the CGMD simulations well reproduced the dynamics of protein-ligand binding. In addition, we calculated flux of the ligand using the trajectories during the periods when the ligand was approaching to the substrate-binding pocket of the protein. We found that the ligand tended to enter the substrate-binding pocket through specific pathways along the grooves on the protein surface.
Negami et al., J. Comput. Chem. 35, 1835 (2014).
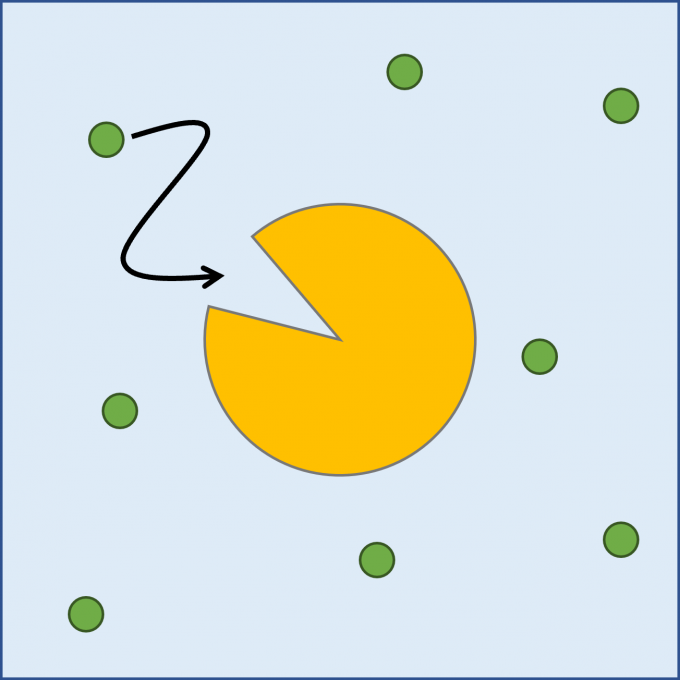
Fig. 1: A schematic representation of a protein-ligand binding process. The protein is shown with a yellow object with a cleft and the ligand molecules are shown with green circles. Cyan background represents the solvent. The ligand moves toward the substrate-binding pocket of the protein represented by the cleft and binds to it.
Fig. 2: The process of the ligand entering the substrate-binding pocket of the protein observed during the CGMD simulation. The ligand molecules are shown with a stick representation. Hydrophilic and hydrophobic regions on the protein surface are colored blue and orange, respectively.
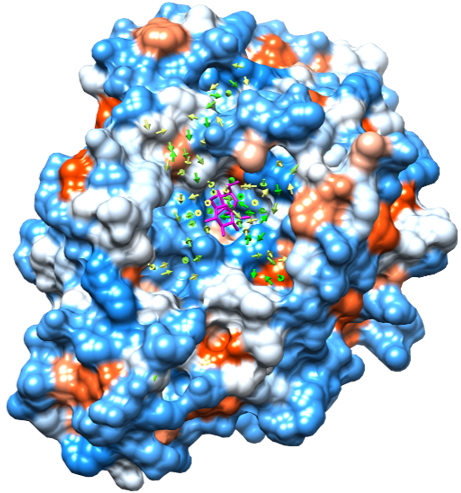
Fig. 3: The flux of the ligand. The protein surface is colored in the same manner as Fig. 2. The flux vectors are shown with yellow to green arrows according to their magnitude. The ligand of the crystal structure is superposed and is shown with a stick representation colored magenta.
Development of a coarse-grained model of a protein that changes its conformation coupled to ligand binding
Some proteins change their conformations upon binding to ligands. In this study, we developed a coarse-grained model of a protein that changes its conformation coupled to ligand binding by combining it with an elastic network model that has energy minima corresponding to the structures before and after the conformational change. There are two types of the conformational change process coupled to ligand binding. One is the “conformational selection” process, in which the protein is in equilibrium between two conformations and the ligand binds to one conformation. The other is the “induced fit” process, in which the conformation changes after the ligand binds to the protein. We showed that the developed model can reproduce both the processes by changing parameters (Fig. 4).
Negami et al., Chem. Phys. Lett. 742, 137144 (2020).
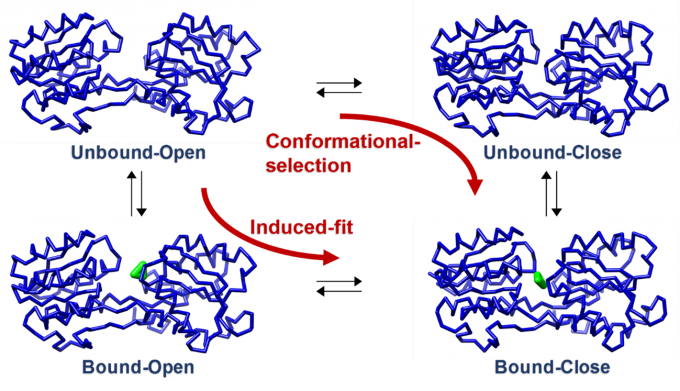
Fig. 4: The coarse-grained model of a protein that changes its conformation coupled to ligand binding. The backbone structure of the protein and the ligand atoms are shown with a blue tube and green spheres, respectively. This protein adopts an open conformation in the ligand-free state and a closed conformation in the ligand-bound state.